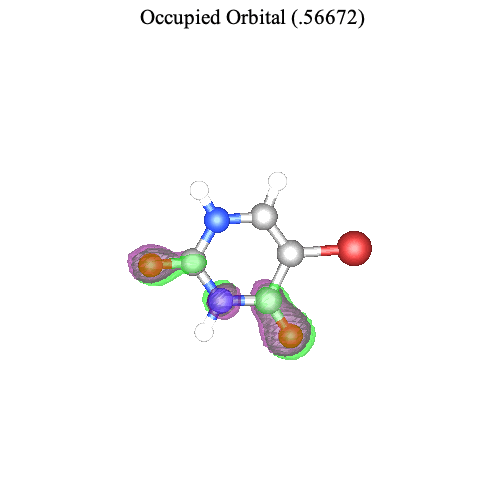
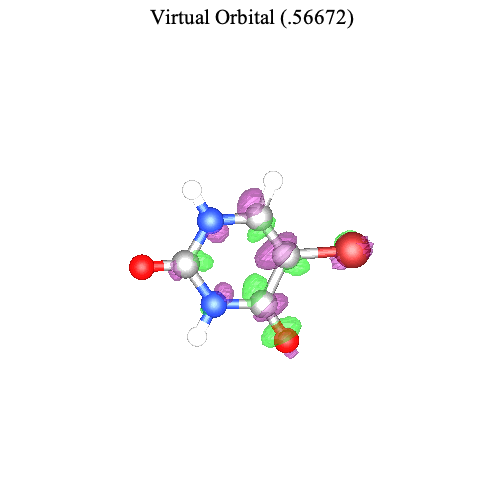
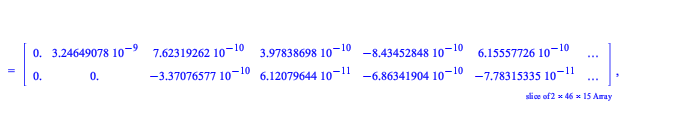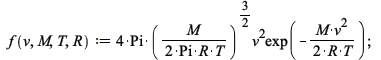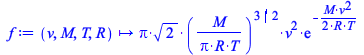Maple
Leistungsfähige intuitive Mathematiksoftware
• Maple für Akademiker • Maple für Studenten • Maple Learn • Maple Calculator App • Maple für Industrie und Behörden • Maple for Individuals
Erweiterungen für Maple
• Books & Studienführer • Maple Toolboxen • MapleNet • Kostenloser Maple-Player
Math Success Platform
Verbesserung der Studienerfolgsquote
Maple Flow
Engineering calculations & documentation
• Maple Flow • Maple Flow Migration Assistant